We will use a simple approach to inspect the mapping of reads to a reference genome. Creating manually reads to map, we will be able to better understand what a mapper can do for us.
Inspecting files
When we need to feed input files or output files, to be sure we are feeding the right files, and finally to check that the output files makes sense.
A mapping file will need as inputs:
- A target reference to map the sequences against (FASTA file)
- One or more (usually millions!) sequences to map (typically FASTQ files, but FASTA is usually allowed)
The output will be a SAM file (or directly a BAM file), which is a text file that contains the alignment of the reads to the reference genome.
How can we inspect a file?
- Use
less -Sto see if the format looks reasonable (orgzip -dc FILE.gz | less -Sif the file is compressed) - Use format specific tools to gather some infos (e.g.
seqfufor sequences,samtoolsfor BAM files)
Creating the new environment
mamba create -n mapping \
-c conda-forge -c bioconda \
bwa samtools seqfu visidata-
bwais a software package for mapping low-divergent sequences against a large reference genome, such as the human genome. -
samtoolsis a suite of programs for interacting with SAM files, including to convert SAM to BAM and viceversa. -
seqfuis a tool to manipulate sequences, including reverse complement, translation, and more. -
visidata is a terminal application for exploring and analyzing tabular data (
vd)
As usual, to be able to use these tools we need to:
conda activate mapping
![]() we used the reference from the learn_bash repository:
we used the reference from the learn_bash repository:
git clone https://github.com/telatin/learn_bash.git
Creating a small dataset
We will use a small dataset to test the mapping. We can systematically create such datasets, but to make an easier start we can do this manually, creating a valid FASTA file
- We open the
vir_genome.fnafile - We copy the first 20 (approximately) lines and save them in a new file.
- Now we can copy the first characters of the sequences to be saved as “Read1”
- We can copy some characters downstream the sequence to be saved as “Read2”
- We can then join two segments of the sequence to create a new sequence “Read3_largegap”, we can insert some characters in the middle of the sequence in lowercase, to remember we added those
and so and forth. When done let’s save the file as small_reads.fasta.
>Read1
GGGCGGCGACCTCGCGGGTTTTCGCTATTTATGAAAATTTTCCGGTTTAAGGCGTTTCCGTTCTTCTTC
>Read2
CTGAAACGGGATATCATCAAAGCCATGAACAAAGCAGCCGCGCTGGATGAACTGATACCGGGGTTGCTGAG
>Read3_largegap
CGAACAGTCAGGTTAACAGGCTGCGGCATTTTGTCGGCTCTCCGACGTTCCTGGGTGACAAGCGTATTGAAG
>Read2_insert
GAAACGGGATATCATCAAAGCCATGAACAAAGCatcgAGCCGCGCTGGATGAACTGATACCGGGGTTGCTGAG
...
All this reads are taken from the reference, in the forward strand. To also check the effect of query sequences in the opposite strand we can append at the bottom of the file the reverse complement of the sequences:
seqfu rc small_reads.fasta >> small_reads_rc.fastaThe final file is ![]() available here.
available here.
Checking our reads
We can use seqfu to check the sequences in the file:
seqfu stats small_reads.fasta# Check how long each read is
seqfu cat --add-len small_reads.fasta | grep ">"Mapping the reads
With bwa we need to create an index of the reference genome:
bwa index path/to/vir_genome.fnaAnd then we can map the reads. bwa output will be a SAM output, that we will redirect to a file.
bwa mem path/to/vir_genome.fna small_reads.fasta > small.samInspecting the SAM file
Let’s start with less -S:
less -S small.samWe notice that there are some header lines starting by @.
These are metadata lines that describe the reference genome and the reads.
In particular you will see one line for each reference sequence (in our case we have only one),
that stores its length:
@SQ SN:NC_001416.1 LN:48502
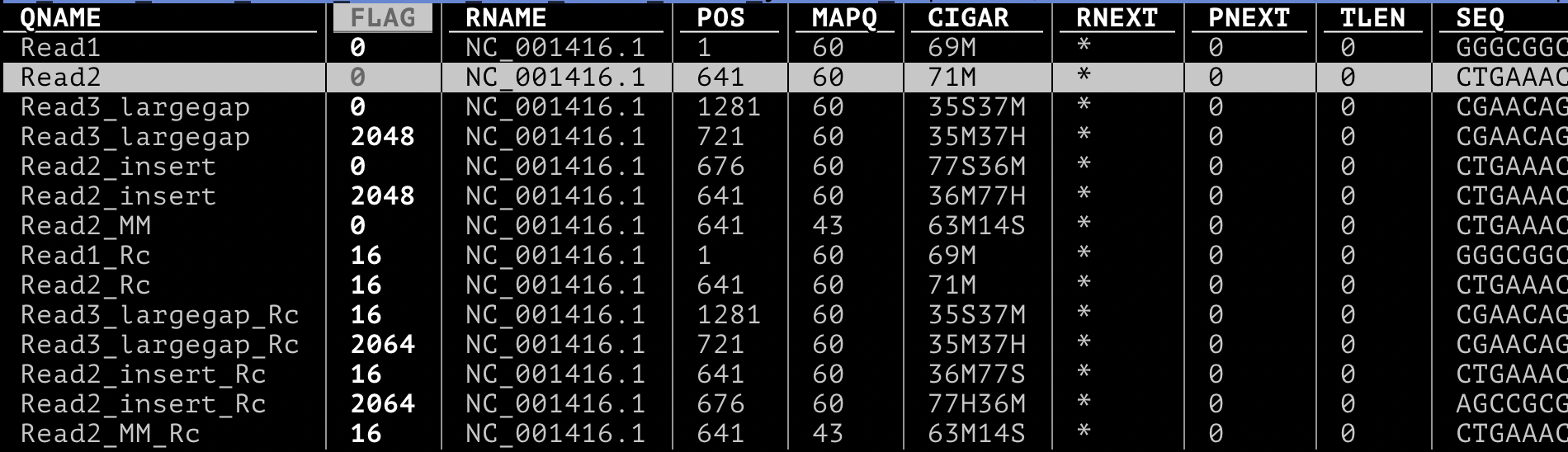
Then we have a tabular output with these columns:
- QNAME: This is the name or identifier of the read.
- FLAG: A bitwise flag that contains information about the alignment. It can tell you if the read is paired, mapped, etc.
- RNAME: The name of the reference sequence where the read aligns.
- POS: The leftmost position of the alignment on the reference sequence (1-based).
- MAPQ: A score indicating the quality of the mapping.
- CIGAR: A string that describes how the read aligns to the reference, including matches, insertions, deletions, etc.
- RNEXT: The reference name of the mate pair (for paired-end reads).
- PNEXT: The position of the mate pair.
- TLEN: The observed length of the template.
- SEQ: The actual sequence of the read.
- QUAL: The quality scores for each base in the read.
To view the main part of the SAM file as a table we can create a header, and then append the content of the sam file (excluding the header!) at the bottom:
echo -e "QNAME\tFLAG\tRNAME\tPOS\tMAPQ\tCIGAR\tRNEXT\tPNEXT\tTLEN\tSEQ\tQUAL" > sam_table.tsv
grep -v "^@" small.sam >> sam_table.tsv
vd sam_table.tsvWe should now see the SAM in a table format, where we can sort the columns, filter, and more.
A brief overview
There’s plenty of material on the SAM format:
To try with a bigger dataset:
We can quickly notice that:
- Each line of the output represent one alignment, and not one read. Read3_largegap, for example, is split in two alignments, as we can expect as we created a chimeric read (this would happen with a spliced read, for example).
- The FLAG is a number that stores a series of attributes. We can notice that all the reverse complement reads have different flags.
- The CIGAR (Compact Idiosyncratic Gapped Alignment Report) string describes how a read aligns to the reference sequence, indicating matches, insertions, deletions, and other alignment features.
From SAM to BAM
Using samtools we can convert the SAM file to a BAM file:
# Convert SAM to BAM: -b means create a "bam" file as output
samtools view -b fake.sam > fake.bamWe can sort the output file by position:
samtools sort -o fake.sorted.bam fake.bam
samtools index fake.sorted.bamAnd then we can get some statistics from the alignment:
samtools flagstat fake.sorted.bam![]() Using the pipe we can directly create a sorted bam:
Using the pipe we can directly create a sorted bam:
bwa mem path/to/vir_genome.fna small_reads.fasta | samtools sort --write-index -o piped.bam -