We converted the tutorial into a very simple Nextflow pipeline. The code is available from Github
What do you need to run
The pipeline will need
- your assembly (FASTA),
- a set of paired end short reads,
- and the databases (for Genomad and CheckV), that now you know you can download with votuderep.
The reads are to be supplied as a mapping file that you can create with votuderep tabulate.
votuderep tabulate ~/virome/reads -o ~/reads.csv
Executing the pipeline
One of the cool things about Nextflow is that you don’t need to download the pipeline from github, and you don’t need to install the dependencies: they will be fetched using your favourite source (in our case, Docker). We could use Nextflow also to download the databases…
nextflow run quadram-institute-bioscience/nf-ebame-virome -profile docker \
--assembly ~/virome/human_gut_assembly.fa.gz \
--reads ~/reads.csv \
--genomad_db ~/db/genomad_db/ \
--checkv_db ~/db/checkv-db-v1.5/ \
--outdir ~/nf-ebame-virome-out
After you hit enter, Nextflow will download the pipeline and start orchestrating the tasks
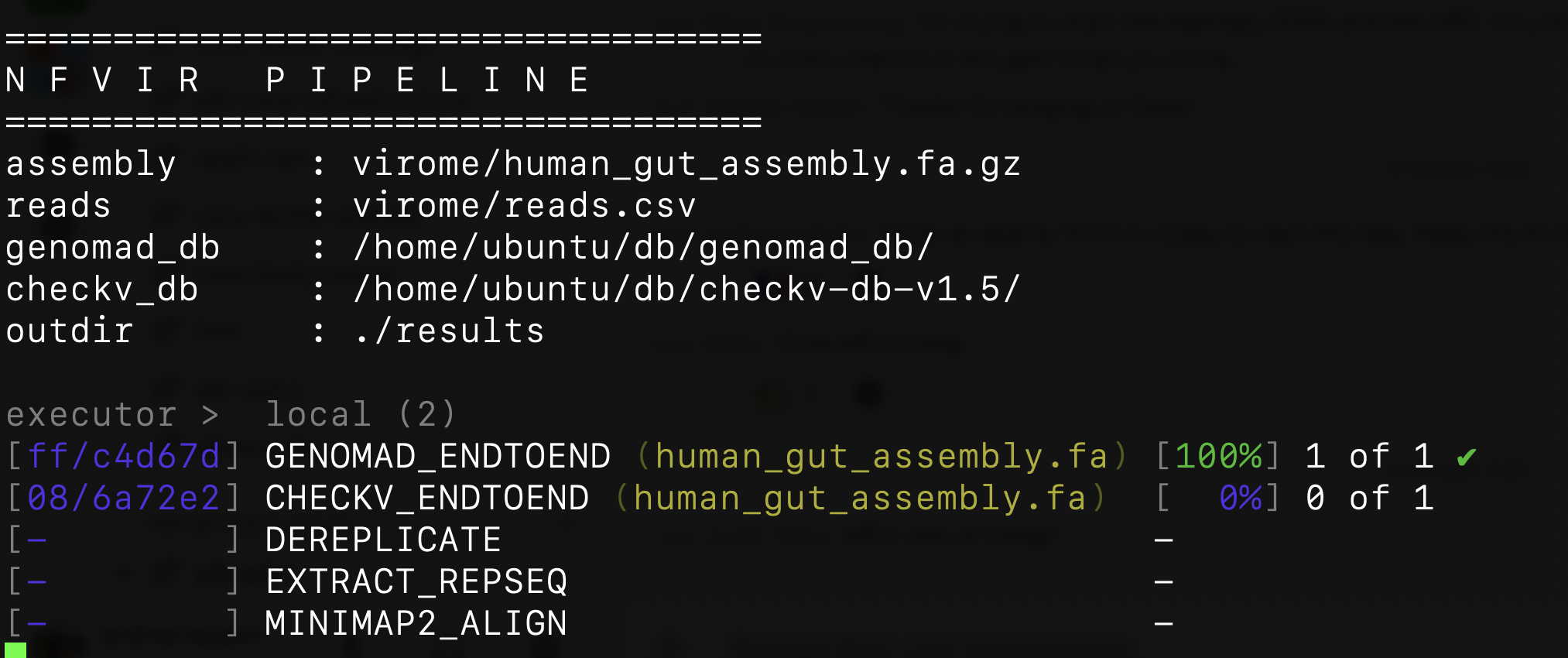
Output
The output folder will contain a subdirectory for each task (it’s still a bit messy):
output-directory
├── genomad
│ └── genomad-out
│ ├── input_assembly_aggregated_classification
│ ├── input_assembly_annotate
│ │ └── input_assembly_mmseqs2
│ │ ├── ...
│ ├── input_assembly_find_proviruses
│ │ └── input_assembly_provirus_mmseqs2
│ │ ├── ...
│ ├── input_assembly_marker_classification
│ ├── input_assembly_nn_classification
│ │ ├── ...
│ └── input_assembly_summary
├── dereplicated-votus (dereplicated vOTU FASTA)
├── checkv
│ └── checkv-out
│ └── tmp...
├── alignments (your BAM files)
└── pipeline_info
Previous submodule:
Workflows