Denoising with DADA2
The core DADA2 algorithm uses the error model learned in the previous chapter to distinguish true biological sequences from sequences that arose through PCR or sequencing errors. It works by asking: given the error model, is this slightly different sequence more likely to be a genuine variant, or an error-derived copy of a more abundant sequence?
The result is a set of Amplicon Sequence Variants (ASVs) : exact sequences with no artificial clustering.
dadaFs_file <- file.path ( path_precomp , "dadaFs.rds" ) dadaRs_file <- file.path ( path_precomp , "dadaRs.rds" ) if ( ! file.exists ( dadaFs_file ) ) { message ( "Denoising forward reads..." )
dadaFs <- dada ( filtFs , err = errF , multithread = TRUE )
saveRDS ( dadaFs , dadaFs_file )
} else { dadaFs <- readRDS ( dadaFs_file )
}
Sample 1 - 13226 reads in 4843 unique sequences.
Sample 2 - 178641 reads in 22804 unique sequences.
Sample 3 - 12109 reads in 4696 unique sequences.
Sample 4 - 1419 reads in 581 unique sequences.
Sample 5 - 34239 reads in 11798 unique sequences.
Sample 6 - 193289 reads in 26735 unique sequences.
Sample 7 - 221122 reads in 21753 unique sequences.
Sample 8 - 150467 reads in 15443 unique sequences.
Sample 9 - 189922 reads in 24078 unique sequences.
Sample 10 - 227640 reads in 22363 unique sequences.
Sample 11 - 210626 reads in 20738 unique sequences.
Sample 12 - 191040 reads in 24153 unique sequences.
Sample 13 - 233210 reads in 29369 unique sequences.
Sample 14 - 107509 reads in 25626 unique sequences.
Sample 15 - 219755 reads in 23609 unique sequences.
Sample 16 - 213873 reads in 22116 unique sequences.
Sample 17 - 230240 reads in 29372 unique sequences.
Sample 18 - 209712 reads in 42650 unique sequences.
Sample 19 - 241630 reads in 45010 unique sequences.
Sample 20 - 209072 reads in 36916 unique sequences.
Sample 21 - 253245 reads in 32445 unique sequences.
Sample 22 - 243119 reads in 43825 unique sequences.
Sample 23 - 238579 reads in 37930 unique sequences.
Sample 24 - 217397 reads in 41176 unique sequences.
Sample 25 - 218668 reads in 39233 unique sequences.
Sample 26 - 239236 reads in 43927 unique sequences.
Sample 27 - 237723 reads in 42642 unique sequences.
Sample 28 - 208982 reads in 43512 unique sequences.
Sample 29 - 256480 reads in 44096 unique sequences.
if ( ! file.exists ( dadaRs_file ) ) { message ( "Denoising reverse reads..." )
dadaRs <- dada ( filtRs , err = errR , multithread = TRUE )
saveRDS ( dadaRs , dadaRs_file )
} else { dadaRs <- readRDS ( dadaRs_file )
}
Sample 1 - 13226 reads in 6970 unique sequences.
Sample 2 - 178641 reads in 28216 unique sequences.
Sample 3 - 12109 reads in 6149 unique sequences.
Sample 4 - 1419 reads in 729 unique sequences.
Sample 5 - 34239 reads in 15345 unique sequences.
Sample 6 - 193289 reads in 32391 unique sequences.
Sample 7 - 221122 reads in 29710 unique sequences.
Sample 8 - 150467 reads in 22351 unique sequences.
Sample 9 - 189922 reads in 30935 unique sequences.
Sample 10 - 227640 reads in 29549 unique sequences.
Sample 11 - 210626 reads in 28342 unique sequences.
Sample 12 - 191040 reads in 33939 unique sequences.
Sample 13 - 233210 reads in 35191 unique sequences.
Sample 14 - 107509 reads in 18547 unique sequences.
Sample 15 - 219755 reads in 31439 unique sequences.
Sample 16 - 213873 reads in 29943 unique sequences.
Sample 17 - 230240 reads in 36340 unique sequences.
Sample 18 - 209712 reads in 53263 unique sequences.
Sample 19 - 241630 reads in 55632 unique sequences.
Sample 20 - 209072 reads in 47285 unique sequences.
Sample 21 - 253245 reads in 41514 unique sequences.
Sample 22 - 243119 reads in 59760 unique sequences.
Sample 23 - 238579 reads in 52820 unique sequences.
Sample 24 - 217397 reads in 51954 unique sequences.
Sample 25 - 218668 reads in 49783 unique sequences.
Sample 26 - 239236 reads in 54978 unique sequences.
Sample 27 - 237723 reads in 49322 unique sequences.
Sample 28 - 208982 reads in 52167 unique sequences.
Sample 29 - 256480 reads in 55448 unique sequences.
What does the dada2 output tell us?
# Inspect the first sample's denoising result dadaFs [[ 1 ] ]
dada-class: object describing DADA2 denoising results
90 sequence variants were inferred from 4843 input unique sequences.
Key parameters: OMEGA_A = 1e-40, OMEGA_C = 1e-40, BAND_SIZE = 16
The output reports how many unique sequences were identified in the sample and how many reads support each one. The ratio of reads to unique sequences gives an indication of how much error correction was performed.
Merging paired-end reads
After denoising, forward and reverse reads are merged to reconstruct the full amplicon sequence. Merging requires a minimum overlap region between the denoised forward and reverse reads — by default, at least 12 bp.
mergers_file <- file.path ( path_precomp , "mergers.rds" ) if ( ! file.exists ( mergers_file ) ) { message ( "Merging paired reads..." )
mergers <- mergePairs ( dadaFs , filtFs , dadaRs , filtRs , verbose = TRUE )
saveRDS ( mergers , mergers_file )
} else { mergers <- readRDS ( mergers_file )
}
If a large proportion of reads fail to merge (visible in the tracking table later), the most common causes are:
Insufficient overlap — increase truncLen for one or both reads, or reduce it if the reads are too short
Primer residues — verify that primers were fully removed before this analysis
Wrong amplicon size — the reads must cover the amplicon with some overlap
Build the sequence table
makeSequenceTable() produces a sample × ASV matrix of read counts. This is the central data object in DADA2.
seqtab <- makeSequenceTable ( mergers ) cat ( "Dimensions (samples × ASVs):" , dim ( seqtab ) , "\n" )
Dimensions (samples × ASVs): 29 18934
Sequence length distribution
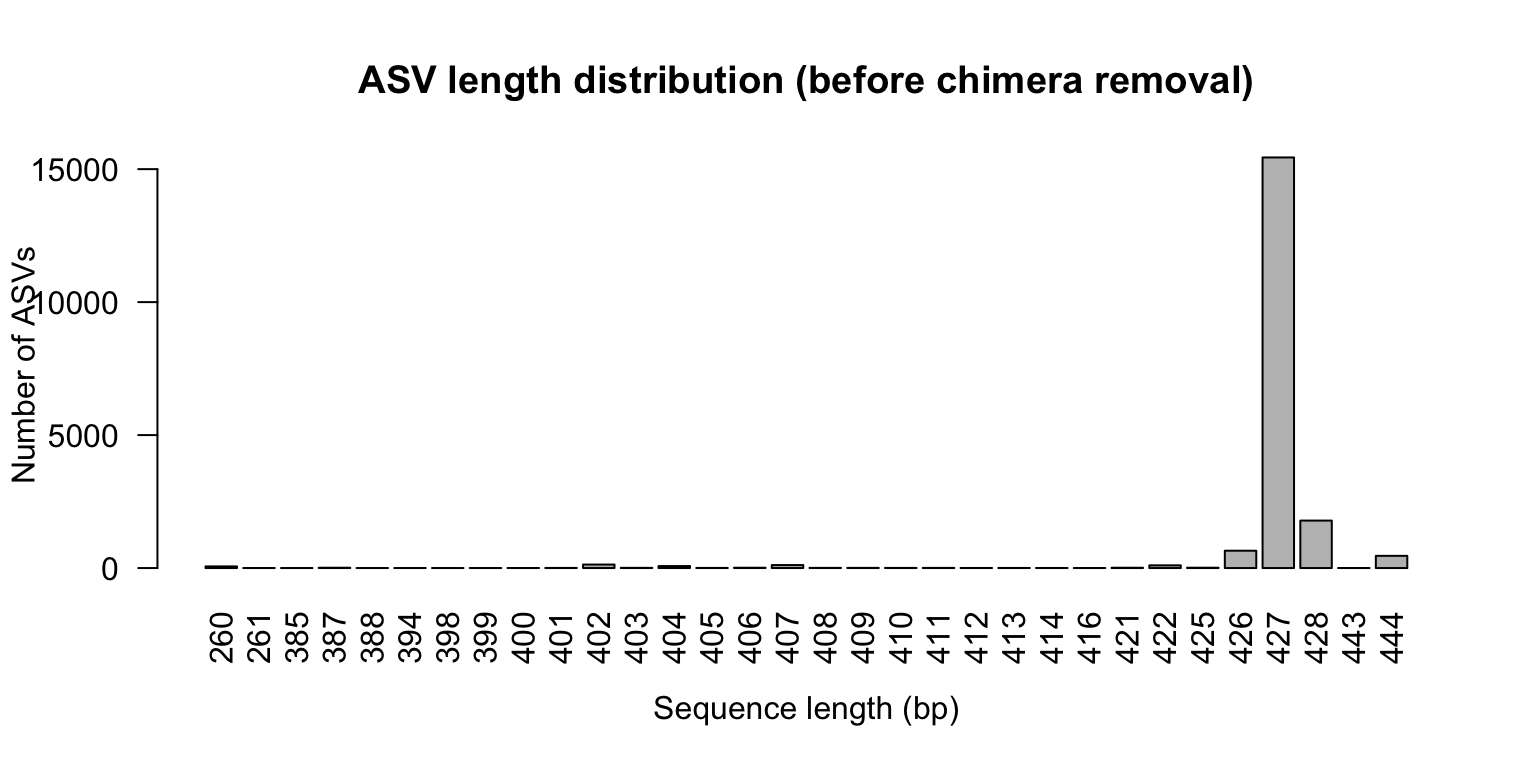
The expected amplicon length for the V3–V4 region is approximately 400–470 bp. Any sequences outside this range are likely artefacts (non-specific amplification or chimeras) and will often be removed in subsequent steps.
260 261 385 387 388 394 398 399 400 401 402 403 404
62 2 1 12 1 1 1 1 2 5 132 8 75
405 406 407 408 409 410 411 412 413 414 416 421 422
5 12 112 7 7 5 6 3 4 3 2 12 102
425 426 427 428 443 444
13 652 15440 1785 2 459
# Visualise barplot ( seq_lengths , xlab = "Sequence length (bp)" ,
ylab = "Number of ASVs" ,
main = "ASV length distribution (before chimera removal)" ,
las = 2 )
Chimera removal
Chimeric sequences are PCR artefacts created when an incomplete extension product from one cycle acts as a primer in a subsequent cycle, fusing the 5′ end of one template with the 3′ end of another. DADA2 identifies chimeras using the removeBimeraDenovo() function, which checks whether each sequence can be reconstructed from two more-abundant “parent” sequences.
seqtab_file <- file.path ( path_precomp , "seqtab_nochim.rds" ) if ( ! file.exists ( seqtab_file ) ) { message ( "Removing chimeras..." )
seqtab.nochim <- removeBimeraDenovo ( seqtab ,
method = "consensus" ,
multithread = TRUE ,
verbose = TRUE )
saveRDS ( seqtab.nochim , seqtab_file )
} else { seqtab.nochim <- readRDS ( seqtab_file )
} cat ( "ASVs before chimera removal:" , ncol ( seqtab ) , "\n" )
ASVs before chimera removal: 18934
cat ( "ASVs after chimera removal:" , ncol ( seqtab.nochim ) , "\n" )
ASVs after chimera removal: 1091
pct_retained <- round ( 100 * sum ( seqtab.nochim ) / sum ( seqtab ) , 1 ) cat ( "Proportion of reads retained after chimera removal:" , pct_retained , "%\n" )
Proportion of reads retained after chimera removal: 89.8 %
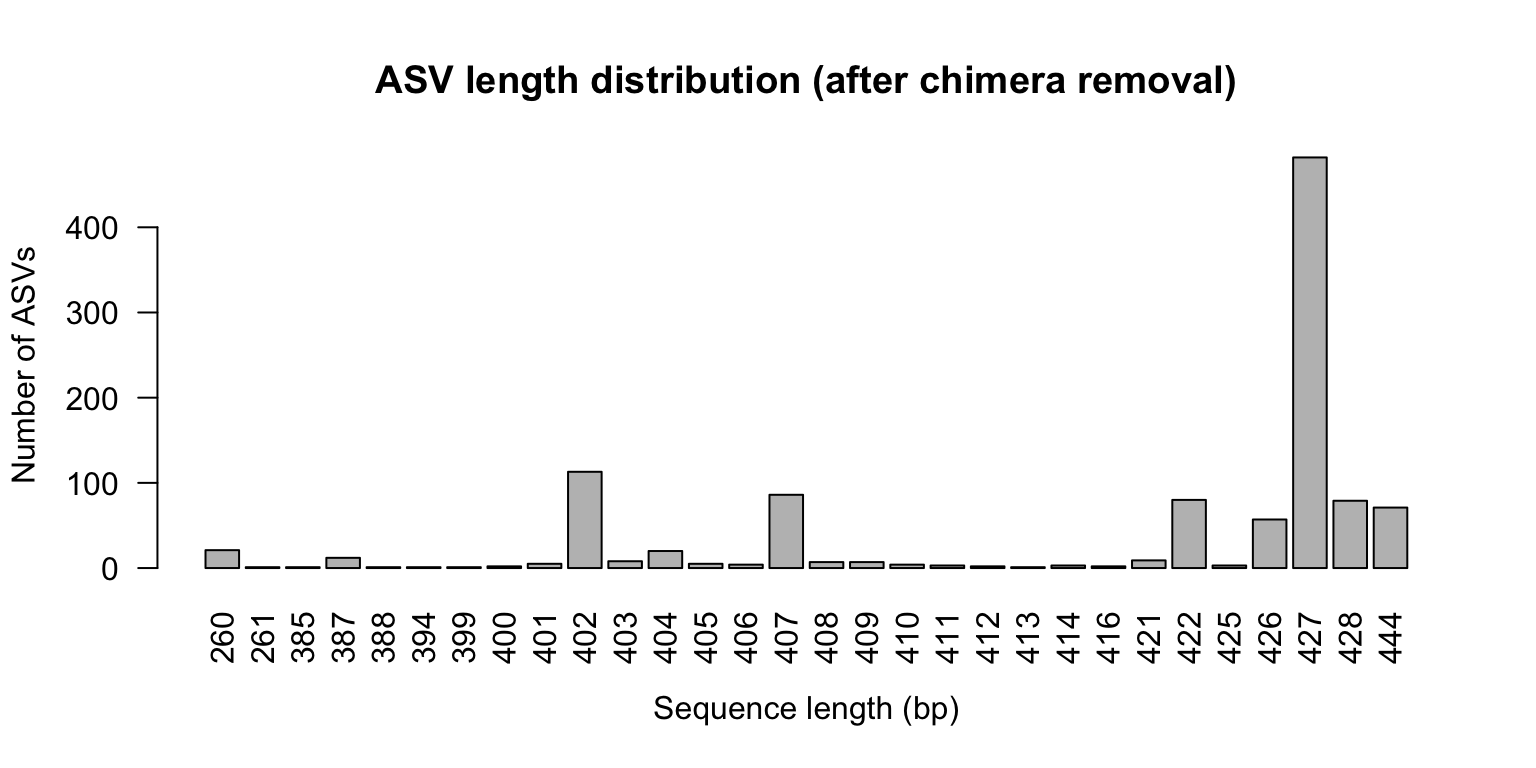
It is normal for a large number of unique sequences to be flagged as chimeric — often 10–30 % of ASVs. However, chimeric reads usually represent a small proportion of total reads (< 10 %), because chimeras are typically rare relative to their parent sequences. If you lose > 20 % of reads here, double-check that primers were fully trimmed.
seq_lengths_nc <- table ( nchar ( getSequences ( seqtab.nochim ) ) ) print ( seq_lengths_nc )
260 261 385 387 388 394 399 400 401 402 403 404 405 406 407 408 409 410 411 412
21 1 1 12 1 1 1 2 5 113 8 20 5 4 86 7 7 4 3 2
413 414 416 421 422 425 426 427 428 444
1 3 2 9 80 3 57 482 79 71
barplot ( seq_lengths_nc , xlab = "Sequence length (bp)" ,
ylab = "Number of ASVs" ,
main = "ASV length distribution (after chimera removal)" ,
las = 2 )
Tracking reads through the pipeline
A key quality-control step is to track how many reads survive each stage of the pipeline. Large losses at any single step point to a specific problem.
getN <- function ( x ) sum ( getUniques ( x ) ) track <- cbind ( out ,
denoisedF = sapply ( dadaFs , getN ) ,
denoisedR = sapply ( dadaRs , getN ) ,
merged = sapply ( mergers , getN ) ,
nonchim = rowSums ( seqtab.nochim )
) colnames ( track ) <- c ( "input" , "filtered" , "denoisedF" , "denoisedR" , "merged" , "nonchim" )
rownames ( track ) <- sample.names write.csv ( track , "dada2_QC.csv" ) track
input filtered denoisedF denoisedR merged nonchim
Commercial_R1 16778 13226 13183 13200 13113 12499
Commercial_R2 218777 178641 177733 178459 176766 176479
Commercial_R3 14993 12109 12049 12098 11922 11670
Control_extraction 2345 1419 1411 1414 1403 1403
Control_fermentation 42709 34239 34114 34105 33515 33162
Lactobacillus_H24_R1 236996 193289 192552 192803 190009 178597
Lactobacillus_H24_R2 271744 221122 220906 220980 220054 217351
Lactobacillus_H24_R3 185402 150467 150220 150328 149791 148672
Lactobacillus_H48_R1 232087 189922 189536 189665 187248 177255
Lactobacillus_H48_R2 277388 227640 227197 227476 226648 222104
Lactobacillus_H48_R3 257945 210626 210217 210457 209083 205846
Lactobacillus_W1_R1 237242 191040 190633 190702 189362 185573
Lactobacillus_W1_R2 283367 233210 232746 232883 230755 224196
Lactobacillus_W1_R3 135637 107509 107352 107386 106982 105252
Lactobacillus_W2_R1 270194 219755 219374 219573 218558 217417
Lactobacillus_W2_R2 262219 213873 213132 213708 212577 209841
Lactobacillus_W2_R3 280448 230240 229271 229942 227359 218116
Spontaneous_H24_R1 260410 209712 208384 208726 197139 162260
Spontaneous_H24_R2 297828 241630 240341 240755 230290 169513
Spontaneous_H24_R3 258467 209072 208224 208561 202745 166417
Spontaneous_H48_R1 311391 253245 252497 252931 229750 203601
Spontaneous_H48_R2 302367 243119 241992 242408 232963 186846
Spontaneous_H48_R3 295072 238579 237584 238146 230008 193478
Spontaneous_W1_R1 266958 217397 216521 216709 206641 165163
Spontaneous_W1_R2 271112 218668 218010 217981 207723 180948
Spontaneous_W1_R3 295075 239236 238162 238617 230440 187702
Spontaneous_W2_R1 291669 237723 236868 237129 225123 190377
Spontaneous_W2_R2 259494 208982 208097 208521 200762 151955
Spontaneous_W2_R3 314666 256480 255722 255872 248160 210209
Visualise read tracking
step_labels <- c ( input = "Raw Input" ,
filtered = "Quality Filtered" ,
denoisedF = "Denoised (Fwd)" ,
denoisedR = "Denoised (Rev)" ,
merged = "Merged Pairs" ,
nonchim = "Non-chimeric"
) track_long <- as.data.frame ( track ) %>% rownames_to_column ( "Sample" ) %>%
pivot_longer ( - Sample , names_to = "Step" , values_to = "Reads" ) %>%
mutate ( Step = factor ( Step , levels = names ( step_labels ) ) )
ggplot ( track_long , aes ( x = Step , y = Reads , fill = Step ) ) + geom_boxplot ( outlier.size = 1 ) +
scale_x_discrete ( labels = step_labels ) +
scale_fill_viridis_d ( ) +
labs ( x = "Pipeline Step" , y = "Number of Reads" ,
title = "Read counts through the DADA2 pipeline" ) +
theme_minimal ( ) +
theme ( axis.text.x = element_text ( angle = 45 , hjust = 1 ) ,
legend.position = "none" )
What to look for:
A gradual, smooth decline across steps is expected and healthy
A sudden large drop at the filtering step → truncation lengths may be too aggressive
A large drop at merging → check overlap between reads; check primers are removed
A large drop at chimera removal → investigate whether primers are truly absent