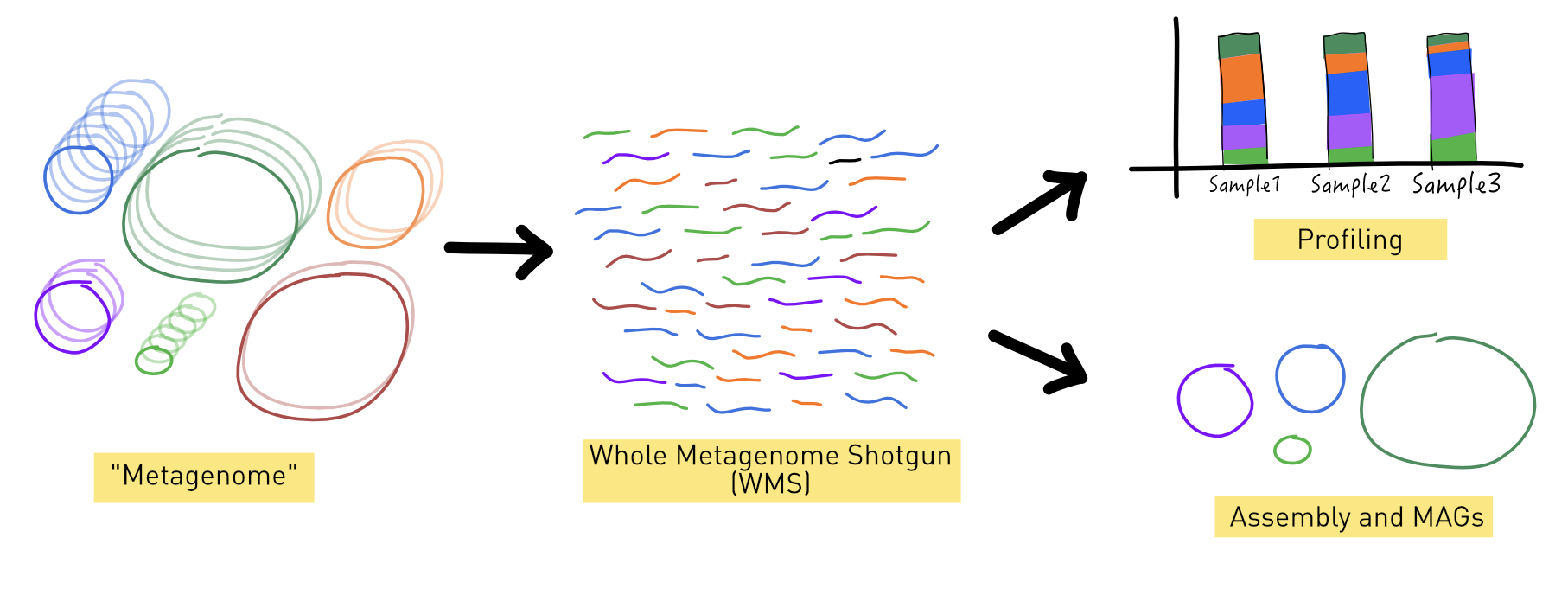
Whole metagenome shotgun
Whole metagenome shotgun (WMS) sequencing involves randomly breaking up DNA from an environmental sample into small fragments, sequencing these fragments, and then using computational methods to reconstruct the original genomes present in the sample.
We will analyse WMS data using two main approaches:
- reads profiling
- de novo assembly
Reads profiling
Reads profiling means trying to assign some information (usually taxonomy, sometimes functions) to each read in the dataset. Then, by aggregating the information from all reads, we can try to infer the composition of the microbial community in the sample.
- MetaPhlAn is a popular tool for taxonomic profiling of metagenomic reads. It focuses on the prokaryotic component of the microbiome (bacteria and archaea), using a database of clade-specific marker genes to provide accurate taxonomic assignments.
- Kraken2 is another widely used tool for taxonomic classification of metagenomic reads. It uses a k-mer based approach to classify reads against a comprehensive database of known sequences, allowing for rapid and sensitive identification of microbial taxa. Several databases are available depending on the memory footprint you want to allocate.
- HUMAnN (The HMP Unified Metabolic Analysis Network) is a tool designed for functional profiling of metagenomic and metatranscriptomic datasets. It aims to identify and quantify the presence of microbial pathways and gene families in a sample, providing insights into the functional potential of the microbial community.
De novo assembly
When assemblying the raw reads, we will obtain a set of contigs - longer sequences that represent (parts of) genomes present in the sample. Each contig represents a fragment of one of the genomes in the sample. The process of binning can then be used to group contigs that likely belong to the same genome, based on sequence composition and coverage patterns. If a “bin” is of sufficient quality and completeness, it can be referred to as a metagenome-assembled genome (MAG).
❓ Do you expect to identify more species with a de novo approach, or via reads profiling?
The MAG (Metagenome Assembled Genome)
Each MAG can be treated as a draft genome produced by a single isolate shotgun, but we have to expect a lower quality due to contamination (contigs coming from other species), reduced completeness (several missing parts).