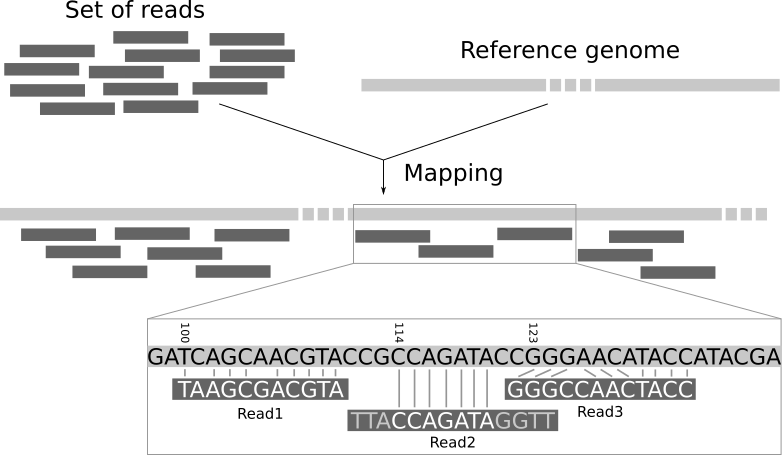
When you have an assembly - that is generated using algorithms that focus on some information of your reads but not all of it (for example k-mer based assemblers will not make use of the full length of the reads, but only of their k-mers) - it is often useful to map back the original reads to the assembly, in order to retrieve information that was not used during the assembly process. This process is called backmapping or reads recruitment.
A key concept when backmapping is the coverage of the contigs by the reads. Coverage is defined as the average number of reads that align to, or “cover” each base position in a contig. High coverage indicates that a contig is well-supported by the original sequencing data, while low coverage may suggest that a contig is less reliable or represents a rare sequence in the sample.
In metagenomics, coverage information is crucial for several reasons:
- Abundance estimation: Coverage can be used to estimate the relative abundance of different organisms in a metagenomic sample. Contigs with higher coverage are likely to originate from more abundant organisms.
- Binning: Coverage patterns across multiple samples can help in binning contigs into putative genomes, as contigs from the same organism are expected to have similar coverage profiles.
- Quality assessment: Coverage can help identify potential assembly errors or chimeric contigs.
Gathering the tools
One of the tools we can use (for both Illumina and long reads) is minimap2. Another popular tool - for shorts reads only - is BWA.
We can use mamba to install it (alongside with another aligner and utilities):
mamba create -n mapping -c conda-forge -c bioconda \
minimap2 bwa samtools bamtocov coverm multiqc
To use the environment you will typically type:
conda activate mapping
Backmapping Illumina reads with Minimap2
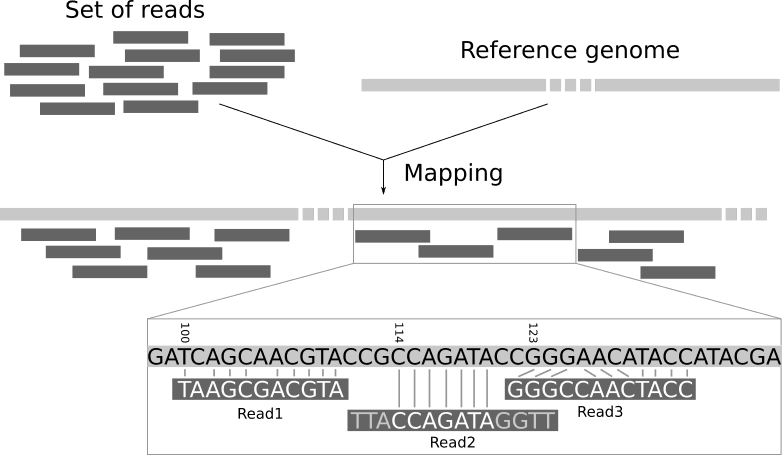
Minimap2 can be used to map Illumina reads to a reference assembly using the -x sr preset for short reads. Here is an example command:
minimap2 -a -t 8 -x sr assembly.fasta reads_R1.fastq.gz reads_R2.fastq.gz > mapped_reads.sam
-
-a: Output in SAM format (default is PAF) -
-t 8: Use 8 threads for parallel processing -
-x sr: Preset for short reads (Illumina)
The SAM format can be converted to a more compact BAM format using samtools, and it’s very common to sort the BAM file (by contig and position) for downstream analyses.
samtools view -bS mapped_reads.sam | samtools sort -@ 4 -o mapped_reads.sorted.bam
samtools index mapped_reads.sorted.bam
-
-b: Output in BAM format (can be omitted in recent versions of samtools), used by samtools view -
-S: Convert SAM to BAM (default is output in SAM), used by samtools view -
-o: Output file name, used by samtools sort -
-@ 4: Use 4 threads for sorting, used by samtools sort
We can map and save to a sorted BAM file in one step like this:
# Map & sort in one step
minimap2 -t 8 -ax sr assembly.fasta reads_R1.fastq.gz reads_R2.fastq.gz | \
samtools view -bS - | samtools sort -@ 4 -o mapped_reads.sorted.bam
# Index the sorted BAM file
samtools index mapped_reads.sorted.bam
Coverage
The coverage is defined as the amount of times a nucleotide is read during the sequencing process. In other words, it indicates how many reads overlap a specific position in the reference sequence. Usually we refer to the average coverage of a contig or genome, which is calculated by averaging the coverage values across all nucleotide positions in that contig or genome.
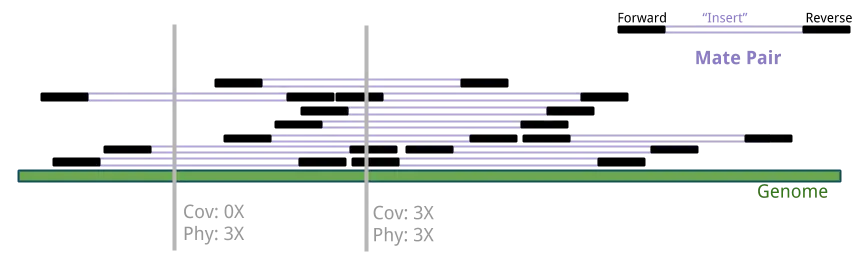
The amount of sequenced nucleotides spanning a base is more specifically called sequence coverage, while the number of fragments (i.e. the two paired-end reads and the insert between them) spanning a base is called physical coverage.
 There are several ways to calculate the coverage from a BAM file. One simple way is to use
There are several ways to calculate the coverage from a BAM file. One simple way is to use samtools depth, which outputs the depth of coverage at each position in the reference.
samtools depth mapped_reads.sorted.bam > coverage.txt
[1] Image source: training.galaxyproject.org
{kind=link}