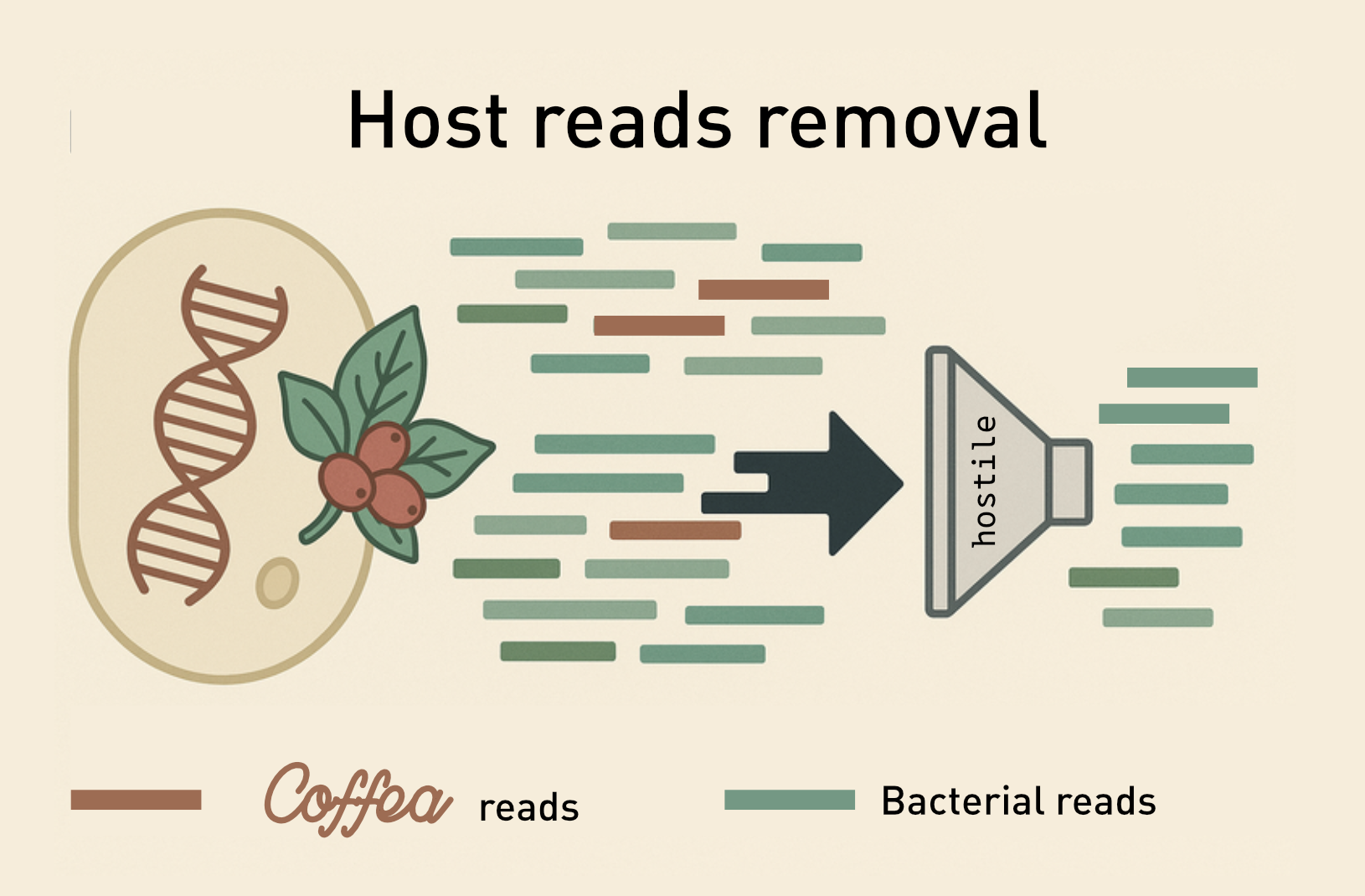
When extracting DNA from samples, if we want to focus on the microbiome we need to get a sample that has as limited contamination from unwanted organisms.
A typical example is the analysis of the gut microbiome: cells from the human host are usually found in the samples (but typically negligible), while for other body sites (e.g skin) the amount of human cells might be vastly superior to the microbiota cells.
In our workshop we won’t focus on how to remove host cells from the samples (but methods exists, and might even introduce further biases)
Removing host reads
There are different approaches, often based on mapping against a reference genome (i.e. the reads mapping are removed). In general for processes like this we should consider:
- Some regions are conserved across genomes, we might remove reads that are actually part of the microbiome (e.g. integrated retroviruses)
- Sensitivity and specificity are traded-off
- Choosing the right reference and eventually masking (removing) low-complexity regions or regions similar to bacterial
So why dont’ we simply leave the host reads?
- The amount of host reads varies across samples, and this would make analyses more complicated trying to compare abundances
- Several methods might classify host reads as bacterial, inducing to major errors especially in low-biomass samples
There is a host that is special: Homo sapiens. In our case we usually want to be more stringent in removing host reads (even at the risk of removing microbiome reads), to avoid patients data being identifiable. The genome is the most private biological property of an organism after all.
Using hostile

Hostile is a pipeline based on alignment tools (bowtie2 or minimap2) to remove reads.
![]() Hostile allows downloading pre-made reference indexes for Human and Mouse reads.
Hostile allows downloading pre-made reference indexes for Human and Mouse reads.
Downloading coffee
You can use NCBI’s datasets tool to download the GCA_036785865.1 reference
as described here.
Index reference (Bowtie)
Note that the FASTA file can be masked to remove regions similar to bacterial’s segments, but we will skip this step for the workshop.
If you will use Bowtie2 (usually recommended for Illumina reads) as aligner, you will need to build an index with this syntax:
bowtie2-build $FASTA_REFERENCE $OUTPUT_INDEX
-
$FASTA_REFERENCEis the path to the FASTA file -
$OUTPUT_INDEXis the path to the output files (basename). You will use this as index for cleaning the reads
Clean reads
The hostile clean workflow will require the FASTQ files to reads (one if single-end, two if paired-end), the index against which aligning the reads and a directory where the output files will be written adding a suffix “clean_1” and “clean_2” respectively.
Redirecting the standard output, we will save a JSON file with the statistics (amount of host reads).
INDEX=/shared/team/2025_training/week5/coffee/coffee-index
OUTDIR=cleaned-reads
mkdir -p $OUTDIR
hostile clean --threads 8 --index $INDEX_PATH \
--fastq1 $R1 --fastq2 $R2 \
--output $OUTDIR/ > stats.json
The flags are self-explanatory, and the variables should be replaced with file paths.
Summary statistics
The output JSON file will contain statistics about the amount of host reads removed.
[
{
"version": "2.0.2",
"aligner": "bowtie2",
"index": "/path/to/db/Coffea_canephora/coffee",
"options": [],
"fastq1_in_name": "ERR2231567_1.fastq.gz",
"fastq1_in_path": "/path/to/ERR2231567_1.fastq.gz",
"reads_in": 16544372,
"reads_out": 11733414,
"reads_removed": 4810958,
"reads_removed_proportion": 0.29079,
"fastq2_in_name": "ERR2231567_2.fastq.gz",
"fastq2_in_path": "/path/to/ERR2231567_2.fastq.gz",
"fastq1_out_name": "ERR2231567_1.clean_1.fastq.gz",
"fastq1_out_path": "2_qc/hostile/ERR2231567_1.clean_1.fastq.gz",
"fastq2_out_name": "ERR2231567_2.clean_2.fastq.gz",
"fastq2_out_path": "2_qc/hostile/ERR2231567_2.clean_2.fastq.gz"
}
]