Hostile is a great tool to remove host reads, and it brings the convenience of pre-built indexes for human and mouse.
It’s important to remember that at its core, Hostile is based on mapping reads to a reference genome, and then removing the mapped reads.
Other approaches exists, for example based on k-mers, or just using an alignment tool directly.
Kraken2
Kraken2 can be used to classify reads against a host genome database, and then separate host from non-host reads. You can find a tutorial on how to build a custom Kraken2 database.
Once you have a Kraken2 database with the host genome, you can use this command to classify and separate reads:
kraken2 --db /path/to/host_db \
--threads 8 \
--unclassified-out clean_#.fastq \
--paired reads_R1.fastq reads_R2.fastq > /dev/null
The # symbol is replaced with _1 and _2 for paired-end reads.
![]() This approach can be fast and memory-efficient, but might be less sensitive than alignment-based methods,
especially if the host genome is not well represented in the database. Also, in the special database used for host removal,
there is only one genome (the host), which might lead to misclassification of reads from conserved regions.
This approach can be fast and memory-efficient, but might be less sensitive than alignment-based methods,
especially if the host genome is not well represented in the database. Also, in the special database used for host removal,
there is only one genome (the host), which might lead to misclassification of reads from conserved regions.
BBMap
Even if we can potentially use any aligner, BBMap is a popular choice for host read removal, possibly because it has a simple method to save unmapped reads directly.
INDIR=...
OUTDIR=cleaned-reads
mkdir -p $OUTDIR/mapped
for R1 in $INDIR/*_1.fastq.gz;
do
R2=${R1/_1/_2}
if [[ ! -e $R2 ]]; then echo Error $R2; exit; fi
BASE=$(basename $R1 | cut -f1 -d_)
bbmap.sh -Xmx48g threads=16 \
path=$BBMAP_INDEX \
in1=$R1 in2=$R2 \
outu1=$OUTDIR/${BASE}_1.fastq.gz \
outu2=$OUTDIR/${BASE}_2.fastq.gz \
outm=$OUTDIR/mapped/${BASE}_host.fastq.gz \
minid=0.95 maxindel=3
done
Comparing the results
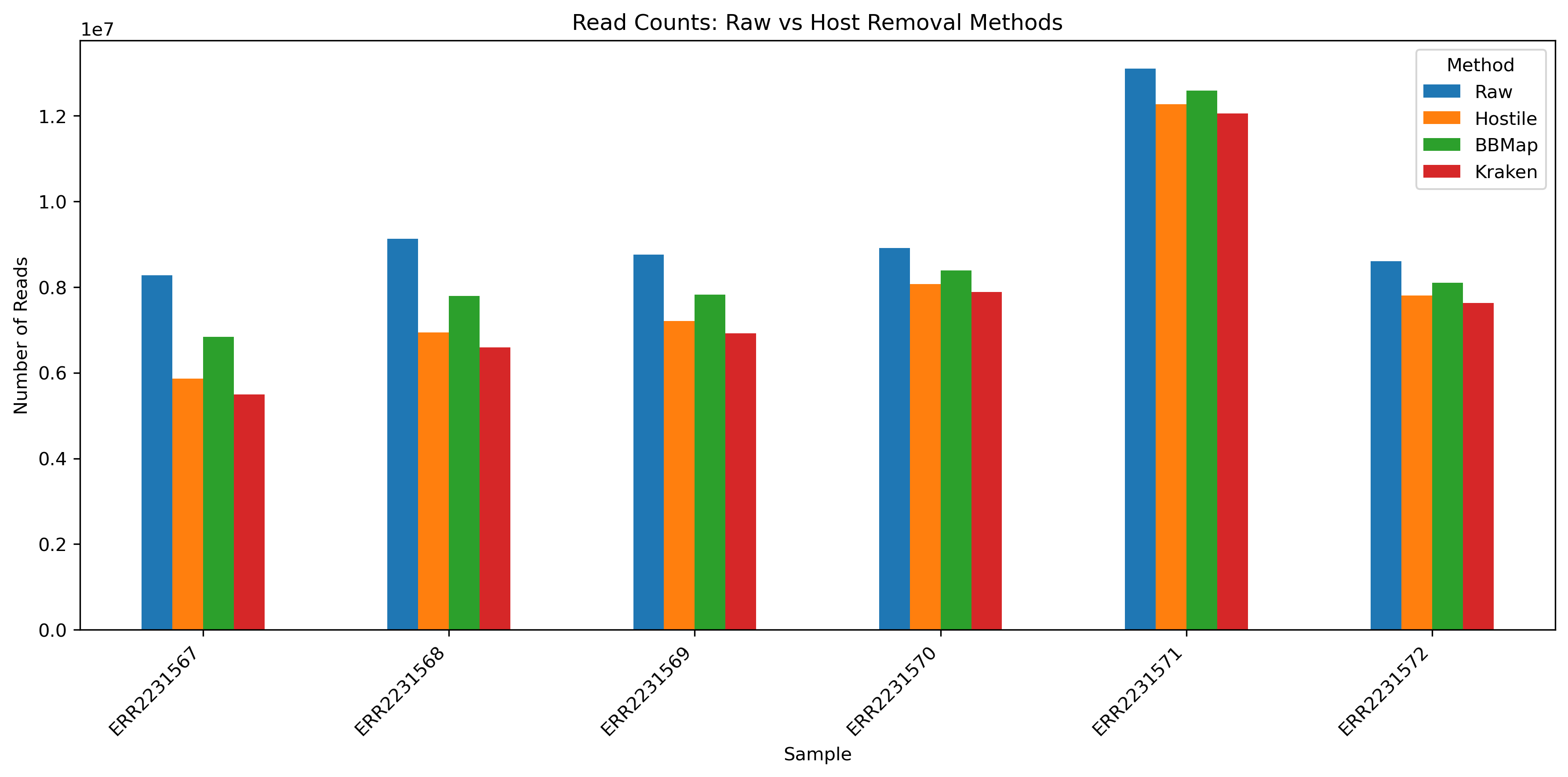
Each tool offers parameters to tweak sensitivity and specificity. In our case we just ran the tools with default or very common parameters.
This Python notebook shows how to plot the number of reads after host removal with different tools.